| Next || Previous || Top |
SURFACE
This keyword data block is used to define the amount and composition of each surface in a surface assemblage. The composition of a surface assemblage can be defined in two ways: (1) implicitly, by specifying that the surface assemblage is in equilibrium with a solution of fixed composition, or (2) explicitly, by defining the amounts of the surfaces in their neutral form (for example, SurfbOH). A surface assemblage may have multiple surfaces and each surface may have multiple binding sites, which are identified by lowercase letters following an underscore. Three types of surfaces are available: DDL (diffuse-double layer) surfaces (Dzombak and Morel, 1990), CD-MUSIC (Charge Distribution MUltiSIte Complexation) surfaces (Hiemstra and Van Riemsdijk, 1996), and non electrostatic surfaces. For DDL and CD-MUSIC surfaces, the composition of the diffuse layer that balances the charged surface can be calculated explicitly (optional). For DDL, the diffuse-layer composition can be calculated by the method of Borkovec and Westall (1983) or by the Donnan approach. For CD-MUSIC, the diffuse-layer composition can be calculated only by the Donnan approach.
Example data block 1
Line 0: SURFACE 1 Surface in equilibrium with solution 10
Line 1: -equilibrate with solution 10
Line 2: Surfa_w 1.0 1000. 0.33
Line 2a: Surfa_s 0.01
Line 2b: Surfb 0.5 1000. 0.33
Line 11: -ddl
Line 0a: SURFACE 2 Explicit diffuse layer
Line 1a: -equilibrate with solution 10
Line 3: -sites_units absolute
Line 2c: Surfa_w 1.0 1000. 0.33
Line 2d: Surfa_s 0.01
Line 2e: Surfb 0.5 1000. 0.33
Line 4: -diffuse_layer 2e-8
Line 0b: SURFACE 3 CD_MUSIC surface with Donnan layer
Line 1b: -equilibrate with solution 10
Line 3a: -sites_units density
Line 5: -cd_music
Line 2f: Goe_uni 3.45 96.8 16.52
Line 2g: Goe_tri 2.7
Line 6: -capacitances 0.98 0.73
Line 7: -Donnan 1e-8
Line 8: -only_counter_ions true
Line 0c: SURFACE 4 Sites related to pure phase and kinetic reactant
Line 1c: -equilibrate with solution 10
Line 9: Surfc_wOH Fe(OH)3(a) equilibrium_phase 0.1 1e5
Line 9a: Surfc_sOH Fe(OH)3(a) equilibrium_phase 0.001
Line 9b: Surfd_sOH Al(OH)3(a) kinetic_reactant 0.001 2e4
Line 10: -no_edl
Line 0d: SURFACE 5 Clay surface with diffusion through Donnan layer
Line 1d: -equilibrate with solution 5
Line 2h: Clay_planar 1.59 37. 1.407e4
Line 2i: Clay_ii 0.01
Line 2j: Clay_fes 0.85e-3
Line 7a: -Donnan 9.6e-10 viscosity 1.0
Line 8a: -only_counter_ions true
Line 0e: SURFACE 6 Clay surface with variable Donnan layer
Line 1e: -equilibrate with solution 6
Line 2k: Clay_planar 1.59 37. 1.407e4
Line 7b: -Donnan debye_lengths 3.4 limit_ddl 0.9 viscosity 1
Line 0f: SURFACE 7 Colloidal Ferrihydrite particles
Line 1f: -equilibrate with solution 7
Line 2l: Hfo_w 2.4e-3 600 1.06 Dw 1e-11
Line 2m: Hfo_s 6e-5
Line 7c: -Donnan 1e-12
Line 0f: SURFACE 8 Constant capacitance model
Line 2m: Ha_aH 3.70E-06 1500 0.010
Line 12: -ccm 3.196
Explanation 1
Line 0:
SURFACE
[
number
] [
description
]
SURFACE
is the keyword for the data block.
number
--A positive number designates the surface assemblage and its composition. A range of numbers may also be given in the form
m-n
, where
m
and
n
are positive integers,
m
is less than
n
, and the two numbers are separated by a hyphen without intervening spaces. Default is 1.
description
--Optional comment that describes the surface assemblage.
Line 1:
-equilibrate
number
-equilibrate
--Indicates that the surface assemblage is defined to be in equilibrium with a given solution composition. Optionally,
equil
,
equilibrate
,
-e
[
quilibrate
],
equilibrium
, or
-e
[
quilibrium
].
number
--Solution number with which the surface assemblage is to be in equilibrium. Any alphabetic characters following the identifier and preceding an integer (“with solution” in Line 1) are ignored.
Line 2:
surface binding site,
(
sites
or
site density
)
, specific_area_per_gram, grams,
[
Dw
coefficient
]
surface binding site
--Name of a surface binding site.
sites
--Total number of sites for this binding site, in moles; applies when
-sites_units
is
absolute
.
site density
--Site density for this binding site, in sites per square nanometer; applies when
-sites_units
is
density
.
specific_area_per_gram
--Specific area of surface, in m
2
/g (square meter per gram). Default is 600 m
2
/g.
grams
--Mass of solid for calculation of surface area, g (gram); surface area is
grams
times
specific_area_per_gram
. Default is 0 g.
Dw
coefficient
--Optional diffusion coefficient for the surface, m
2
/s; applies only when
-multi_D
is true in a TRANSPORT calculation. If coefficient > 0, the surface is transported as a colloid with advective, dispersive, and diffusive transport. Default is 0 m
2
/s, which means the surface is immobile.
Line 3:
-sites_units
(
absolute
or
density
)
-sites_units
--Identifier specifies the units for the sites definition.
Absolute
indicates the number of surface sites is given in moles;
density
indicates that the site density is given in sites per square nanometer of surface area. The choice of units applies to all surfaces in the surface assemblage. Default is
absolute
if
-sites_units
is not included. Optionally,
sites_units
,
site_units
,
-s
[
ite_units
], or
-s
[
ites_units
].
absolute
or
density
--
Absolute
indicates the number of sites is given in moles;
density
indicates the site density is given and the number of sites is calculated from the site density and the surface area.
Line 4:
-diffuse_layer
[
thickness
]
-diffuse_layer
--Indicates that the composition of the diffuse layer will be calculated, such that the net surface charge plus the net charge in the diffuse layer will sum to zero. See Notes 1following this section. Either
-diffuse_layer
or
-Donnan
is necessary to calculate the explicit diffuse-layer composition that counterbalances the surface charge. The identifiers
-diffuse_layer
,
-Donnan
, and
-no_edl
are mutually exclusive and apply to all surfaces in the surface assemblage. The
-diffuse_layer
option is not available when using a CD-MUSIC surface (
-cd_music
). Optionally,
diffuse_layer
or
-d
[
iffuse_layer
].
thickness
--Thickness of the diffuse layer, m (meter). Default is 10
-8
m (equals 100 angstrom).
Line 5:
-cd_music
-cd_music
--Indicates that the surfaces in the surface assemblage are CD-MUSIC surfaces. See Notes 1 for using diffuse double layer and
-no_edl
surfaces in a CD-MUSIC surface assemblage. Optionally,
cd_music
or
-cd
[
_music
].
Line 6:
-capacitances
c
1
,
c
2
-capacitances
--Identifier specifies the capacitances for the CD-MUSIC surface. Different surfaces within the surface assemblage may have different capacitances. This option has effect only when
-cd_music
is defined. Defaults are
c
1
= 1 and
c
2
= 5 F/m
2
(farad per square meter) if
-capacitances
is not included. Optionally,
capacitances
or
-ca
[
pacitances
].
c
1
--Capacitance for the 0-1 plane in the CD-MUSIC formulation, F/m
2
.
c
2
--Capacitance for the 1-2 plane in the CD-MUSIC formulation, F/m
2
.
Line 7:
-Donnan
[(
thickness
or
debye_lengths
lengths
[
limit_ddl
limit
])] [
viscosity
fraction
]
-Donnan
--Indicates that the Donnan approach will be used to calculate the composition of the diffuse layer. The identifiers
-diffuse_layer
,
-Donnan
, and
-no_edl
are mutually exclusive and apply to all surfaces in the surface assemblage. The
-Donnan
option is available when using diffuse-double-layer (default) or CD-MUSIC (
-cd_music
) surfaces. Optionally,
Donnan
or
-Do
[
nnan
] (as with all identifiers, case insensitive).
thickness
--Thickness of the diffuse layer in meters. Default is 10
-8
m.
debye_lengths
lengths
--Either
thickness
or
debye_lengths
may be used to define the thickness of the diffuse layer. If
debye_lengths
is used, the Debye length is calculated from the ionic strength of the solution. The thickness of the diffuse double layer is calculated by the product of
lengths
times the Debye length (Appelo and Wersin, 2007).
limit_ddl
limit
--If
debye_lengths
is specified, then, optionally, the amount of water contained in the diffuse layer can be limited.
Limit
is the fraction of the total water (pore space plus diffuse double layer water) that can be in the diffuse double layer. Default for
limit
is 0.8.
viscosity
fraction
--When considering multicomponent diffusion in a TRANSPORT calculation (
-multi_D
true),
fraction
affects the diffusion of ions in the diffuse layer.
Fraction
is the viscosity in the diffuse layer relative to the viscosity in the free pore space. Default is 1.0.
Line 8:
-only_counter_ions
[(
True
or
False
)]
-only_counter_ions
--Indicates that the surface charge will be counterbalanced in the diffuse layer with counter-ions only (the sign of charge of counter-ions is opposite to the surface charge). This option has effect only when
-diffuse_layer
or
-Donnan
is defined. When
-only_counter_ions
is true and
-diffuse_layer
is used, charge balance by co-ion exclusion is neglected (co-ions have the same sign of charge as the surface), meaning that co-ions have the same concentration in the diffuse layer as in the free pore space. When
-only_counter_ions
is true and
-Donnan
is used, co-ions are completely excluded from the diffuse layer. See Notes 1 following this section. Default is
false
if
-only_counter_ions
is not included. Optionally,
only_counter_ions
or
-o
[
nly_counter_ions
].
(True
or
False)
--
True
indicates that the surface charge will be balanced by a surplus of counter-ions in the diffuse layer;
false
indicates that surface charge will be balanced by a counter-ion surplus and a co-ion deficit in the diffuse layer relative to the bulk solution. Optionally,
t
[
rue
] or
f
[
alse
].
Line 9:
surface binding-site formula, name,
[(
equilibrium_phase
or
kinetic_reactant
)]
, sites_per_mole, specific_area_per_mole
surface binding-site formula
--Formula for surface species including stoichiometry of surface site and other surface-complexed elements connected with a pure phase or kinetic reactant. The formula must be charge balanced and is normally the OH-form of the surface binding site. If no elements other than the surface site are included in the formula, then the surface site must be uncharged. If elements are included in the formula and the surface is reacted with a solution, then these elements, in proportion to the mineral present, will be available to desorb and possibly be incorporated in other solids in the system. Further, if the mineral or kinetic reactant is dissolved, these elements will be removed from the solution and (or) other solids in the system in proportion to the mineral or kinetic reactant dissolution.
name
--Name of the pure phase or kinetic reactant that has this kind of surface site. If
name
is the name of a phase, the moles of the phase in the EQUILIBRIUM_PHASES data block with the same number as this surface number (4 for Lines 9 and 9a) will be used to determine the number of moles of surface sites (moles of phase times
sites_per_mole
equals moles of surface sites). If
name
is the rate name for a kinetic reactant, then the moles of the reactant in the KINETICS data block with the same number as this surface number (4 for line 9b) will be used to determine the number of surface sites (moles of kinetic reactant times
sites_per_mole
equals moles of surface sites). Note that the stoichiometry of the phase or reactant must contain sufficient amounts of the elements in the surface complexes defined in Line 3. In the Example data block 1, there must be at least 0.101 mol of oxygen and hydrogen per mole of Fe(OH)3(a).
equilibrium_phase
or
kinetic_reactant
--If
equilibrium_phase
is used, the
name
on the line is a phase defined in an EQUILIBRIUM_PHASES data block. If
kinetic_reactant
is used, the name on the line is the rate name for a kinetic reactant defined in a KINETICS data block. Default is
equilibrium_phase
. Optionally,
e
or
k
; only the first letter is checked.
sites_per_mole
--Moles of surface sites per mole of phase or kinetic reactant, unitless (mol/mol).
specific_area_per_mole
--Specific area of surface, in m
2
/mol (square meter per mole) of equilibrium phase or kinetic reactant. Default is 0 m
2
/mol.
Line 10:
-no_edl
-no_edl
--Indicates that no electrostatic terms will be used in the mass-action equations for surface species and no explicit calculation of the diffuse-layer composition is performed. The identifiers
-no_edl
,
-diffuse_layer
, and
-Donnan
are mutually exclusive and apply to all surfaces in the surface assemblage. Optionally,
no_edl
,
-n
[
o_edl
],
no_electrostatic
,
-n
[
o_electrostatic
].
Line 11: -ddl
-ddl--Indicates that the diffuse double layer model will be used for all surfaces defined in this SURFACE data block. The diffuse double layer model is the default and will be used if neither -cd_music nor -no_edl is defined. Optionally, ddl, -dd[l].
Line 12: -ccm c
-ccm--Indicates that the surfaces in the surface assemblage use the constant-capacitance model. See Notes 1 for using diffuse double layer and -no_edl surfaces with constant capacitance surfaces. Different capacitances can be defined for different surfaces by using multiple line 11. Optionally, ccm, -cc[m], constant_capacitance, or -co[nstant_capacitance].
c--Capacitance in F/m2 (farad per square meter).
Notes 1
The databases included with PHREEQC contain thermodynamic data for a diffuse-double-layer surface named “Hfo” (Hydrous ferric oxide) that are derived from Dzombak and Morel (1990). Two sites are defined for this surface: a strong binding site, Hfo_s, and a weak binding site, Hfo_w. Note that Dzombak and Morel (1990) used 0.2 mol weak sites and 0.005 mol strong sites per mol Fe, a surface area of 5.33
×
10
4
m
2
/mol Fe, and a gram-formula weight of 89 g Hfo/mol Fe; to be consistent with their model, the relative number of strong and weak sites should remain constant as the total number of sites varies. To facilitate consistency, the identifier
-sites_units
density
can be used, which calculates the number of sites from the site density (sites per square nanometer), the specific surface area (square meter per gram), and the mass (grams).
A surface assemblage can have multiple surfaces, each of which can have multiple site types.
SURFACE
1 in Example data block 1 has two surfaces, Surfa and Surfb. Surfa has two binding sites, Surfa_w and Surfa_s; they share the same surface area and have the same electrostatic potential. The link between the two is indicated by the shared surface name, which is followed by an underscore, “_”, and other letter(s) to distinguish the two types of sites. The surface area and mass for Surfa must be defined in the input data for at least one of the two binding sites. Surfb has only one kind of binding site and the area and mass must be defined as part of the input for the single binding site.
SURFACE
2 is the same as
SURFACE
1 except that an explicit calculation of the composition of the diffuse layer is specified. The identifier
-sites_units absolute
is equivalent to the default that is used in
SURFACE
1.
SURFACE
3 defines a CD-MUSIC surface. The data are based on a description of binding on goethite (Hiemstra and Van Riemsdijk, 1996; Rahnemaie and others, 2006) that has two site types (Goe_uni and Goe_tri). The identifiers
-ddl,
-equilibrate
,
-diffuse_layer
,
-sites_units
,
-cd_music
,
-Donnan
,
-only_counter_ions
, and
-no_edl
apply to all surfaces and site types in the surface assemblage. The identifier
-capacitances
is defined for each individual CD-MUSIC surface and does not apply to the entire surface assemblage. A combination of CD-MUSIC, diffuse double layer, and
-no_edl
surfaces cannot be used directly in a
SURFACE
assemblage. However, a diffuse-double-layer surface, like Surfa in
SURFACE
1, will keep its properties in a CD-MUSIC surface assemblage when defined with very high capacitances; for example,
-capacitances
1e5 1e5. Similarly, a
-no_edl
surface will keep its properties in a diffuse-double-layer surface if the surface area is very large (1
×
10
10
m
2
[square meter] for the product of
specific_area_per_gram
and
grams
). By extension, a
-no_edl
surface will keep its properties when defined in a CD_MUSIC surface assemblage if the surface area and capacitances are large. Thus, with these special definitions,
-cd_music
can be used to model simultaneously all of the available types of surfaces (
-no_edl
, diffuse double layer, and CD-MUSIC). If a
-no_edl
surface with a large surface area is included in an assemblage together with identifier
-Donnan
, the thickness may have to be set
to a small number
to avoid the situation where the volume of EDL water becomes unrealistically large.
SURFACE
4 has one surface, Surfc, which has two binding sites, Surfc_w and Surfc_s. The number of binding sites for these two kinds of sites is determined by the amount of Fe(OH)3(a) in EQUILIBRIUM_PHASES 4, where 4 is the same number as the surface number. If
m
represents the moles of Fe(OH)3(a) in EQUILIBRIUM_PHASES 4, then the number of sites of Surfc_w is 0.1
m (
mol) and of Surfc_s is 0.001
m
(mol). The surface area for Surfc is defined relative to the moles of Fe(OH)3(a), such that the surface area is 100,000
m
(m
2
). During batch-reaction simulations the moles of Fe(OH)3(a) in EQUILIBRIUM_PHASES 4 may change, in which case the number of sites of Surfc will change as will the surface area associated with Surfc. Whenever Fe(OH)3(a) precipitates, the specified amounts of Surfc_wOH and Surfc_sOH are formed and all the species that are surface-complexed and in the electrical double layer will be taken from the elements in the system. These formulas are charge balanced and the OH groups are part of the formula for Fe(OH)3(a). The OH is not used in the initial surface-composition calculation, but is critical when amounts of Fe(OH)3(a) vary. Erroneous results will occur if the formula is not charge balanced, and a warning message will be printed if the elements in the surface complex (other than the surface site itself) are not contained in sufficient quantities in the equilibrium phase or kinetic formula.
The number of sites of Surfd in
SURFACE
4 is determined by the amount of a kinetic reactant defined in KINETICS 4, where 4 is the same number as the surface number. Sites related to a kinetic reactant are exactly analogous to sites related to an equilibrium phase. The same restrictions apply--the formula must be charge balanced, and the elements in the surface complex (other than the surface site itself) should be included in the formula of the reactant.
SURFACE
5 is a template for modeling sorption and diffusion in clay rocks; in this case, the Opalinus Clay at Mont Terri in Switzerland (Appelo and others, 2010). A rock dry density of 2.7 kg/L, an overall porosity of 0.161, of which half is accessible for Cl-, a specific surface area of 37 m2/g, and an exchange capacity of 0.114 eq/kg (equivalent per kilogram) are translated to a surface that is defined relative to 1 L of pore water. Thus, 1 L of pore water is in contact with 2.7
×
(1 - 0.161) / 0.161 = 14.07 kg rock. The surface has “clay_planar” sites that express the bulk exchange capacity of the rock (1.59 mol sites). The measured sorption isotherm for Cs+ indicates the presence of two sites: “clay_ii” sites with an intermediate strength for binding Cs+, and “clay_fes” sites on the frayed edges of illite with a very strong and very specific binding strength. The number of these sites and the complexation constants for Cs+ and other cations are obtained by fitting the isotherm, while accounting for two sorption types: one type is for surface complexation, which is specific for the various ions; the other type is connected with charge compensation in the Donnan space, which is in principle, the same for all equal-charged ions (although it can be varied with
-erm_DDL
in keyword SOLUTION_SPECIES to match observed differences). In
SURFACE
5, the exclusion of Cl- is modeled with a Donnan pore space that contains only counter-ions (is Cl- free). Thus, it holds 0.5 L water and has a thickness (derived from this volume and the total area) of 0.5
×
10
-3
/ (37
×
14.07
×
10
3
) = 9.6
×
10
-10
m. This thickness equals 1.9 Debye lengths at the ionic strength of the pore water of 0.368, which is in good agreement with anion-exclusion theory (Schofield, 1947; Tournassat and Appelo, 2011).
An anion-free Donnan pore space is the simplest option and is in line with traditional calculations in soil science. Perhaps more realistically, the anion exclusion can be modeled with
-only_counter_ions
false and an increased thickness of the Donnan layer. The fraction of free (uncharged) pore water, ffree, follows from the Cl- accessible pore space and the potential in the Donnan space, ψD (V):
 , (6)
, (6)
where ε
a
is the accessible porosity (unitless), ε
tot
is the total porosity (unitless), F is the Faraday constant (96485 JV-1eq-1), R is the gas constant (8.314 JK-1mol-1), and T is the temperature (K). The potential in the Donnan space depends on the water composition and the surface charge, while the latter is also a function of the surface complexation constants: higher constants increase complexation and, usually, decrease the surface charge, provided the complexes are uncharged (charged complexes could increase the surface charge). By fixing the complexation constant for Na+ to -0.7, and the constants for the other major cations by matching the measured distribution coefficients in Opalinus Clay, the surface charge--that is, the part of the exchange capacity that is compensated in the Donnan pore space--can be calculated to be 45 percent (Appelo and others, 2010). This results in ffree = 0.117 and thus, 0.883
×
10
-3
m
3
(cubic meter) water in the Donnan pore space per m
3
pore water. Accordingly, the thickness of the Donnan space becomes
0.883
×
10
-3
/ (37
×
14.07
×
10
3
) = 1.7
×
10
-9
m, or 3.4 Debye lengths.
SURFACE
6 illustrates the option to set the thickness of the Donnan layer to be a number of Debye lengths, κ
−1
, given by  (m), where
ε
r
is the relative dielectric constant of water, T is the temperature (kelvin), and I is the ionic strength. Thus, for
SURFACE
5, with
-only_counter_ions
true, the thickness can be defined to be
-Donnan debye_lengths
1.9, while with
-only_counter_ions false
(the default option) the thickness is defined to be
-Donnan debye_lengths
3.4 as in
SURFACE
6. The thickness will now vary with the ionic strength of the solution, and the fraction of free pore water will be adjusted to maintain the same total amount of water. For program convergence, the fraction of Donnan water is limited with
limit_ddl
0.9 in
SURFACE
6 (default is
limit_ddl
0.8).
(m), where
ε
r
is the relative dielectric constant of water, T is the temperature (kelvin), and I is the ionic strength. Thus, for
SURFACE
5, with
-only_counter_ions
true, the thickness can be defined to be
-Donnan debye_lengths
1.9, while with
-only_counter_ions false
(the default option) the thickness is defined to be
-Donnan debye_lengths
3.4 as in
SURFACE
6. The thickness will now vary with the ionic strength of the solution, and the fraction of free pore water will be adjusted to maintain the same total amount of water. For program convergence, the fraction of Donnan water is limited with
limit_ddl
0.9 in
SURFACE
6 (default is
limit_ddl
0.8).
The chemical and physical properties of clay rocks can be measured precisely with diffusion experiments, and PHREEQC can model the experiments by calculating multicomponent diffusion with option
-multi_D
in keyword TRANSPORT. With this option, diffusion is calculated separately for the free (uncharged) pore water and the Donnan pore water, while each solute species has its own diffusion coefficient. It is probable that the electrostatic double layer, mimicked by the Donnan pore space in PHREEQC, has properties that are different from free pore water. The dielectric permittivity is lower in an electrostatic field, which will enhance the complexation of charged ions into neutral species. Such complexation will diminish anion exclusion, and will be different for different anions, depending on charge and hydration radius. This effect can be modeled by defining an enrichment factor in the Donnan pore space with
-erm_ddl
in keyword SOLUTION_SPECIES. Furthermore, the viscosity may be higher in the Donnan pore water than in ordinary water, and diffusion would be lower in proportion with the viscosity ratio. PHREEQC allows setting the viscosity ratio as illustrated in
SURFACE
5 and
SURFACE
6. (Default is 1.0)
SURFACE
7 defines a diffusion coefficient of 10-11 m2/s for a surface consisting of ferrihydrite particles (Hfo in the database). When the diffusion coefficient is larger than 0, the surface will advect and disperse like a normal solute species in a column defined with keyword TRANSPORT and
-multi_D
true, and it will diffuse in accordance with the diffusion coefficient. The surface must be neutral, either charge-free by itself, or by adding a Donnan layer that neutralizes the surface charge. In
SURFACE
7, the Donnan layer is given a small thickness of only 1 picometer to avoid significant variation in water contents in the cells during transport. The transported surfaces will carry the elements in the surface complexed species, as well as the solutes in the Donnan layer. The diffusion coefficient of the surface, either the whole, or part of it, can be modified with the special Basic function CHANGE_SURF. If changed to 0 m2/s, the surface becomes immobile. Thus,
SURFACE
7 is a colloidal particle that can transport heavy metals complexed on its surface while the diffusion coefficient is greater than zero, and it can coagulate with other particles and be deposited depending on chemical or physical conditions in the column by setting the diffusion coefficient to zero with CHANGE_SURF. An example is given at http://www.hydrochemistry.eu/exmpls/colloid.html (accessed June 25, 2012).
Line 1 requires the program to make a calculation to determine the composition of a surface assemblage, termed an “initial surface calculation”. Before any batch-reaction or transport calculations, initial surface calculations are performed to determine the composition of the surface assemblages that would exist in equilibrium with the specified solution (solution 10 for
SURFACE
1 in this Example data block). The composition of the solution will not change during these calculations. In contrast, during a batch-reaction calculation, when a surface assemblage (defined as in Example data block 1 or Example data block 2 of this section) is reacted with a solution with which it is not in equilibrium, both the surface composition and the solution composition will be adjusted to a new equilibrium.
When
-diffuse_layer
or
-Donnan
is not used (default), any charge that develops on the surface during a reaction step will be accompanied by an equal, but opposite, charge imbalance for the solution. Thus, charge imbalances accumulate in the solution and on the surface when surfaces and solutions are separated. This handling of charge imbalances for surfaces is physically incorrect. Consider the following example, where a charge-balanced surface is brought together with a charge-balanced solution. Assume a positive charge develops at the surface. Now remove the surface from the solution. With the default formulation, a positive charge imbalance is associated with the surface,
Z
s
, and a negative charge imbalance,
Z
soln
, is associated with the solution. In reality, the charged surface plus the diffuse layer surrounding it is electrically neutral and the combination should be removed. This would leave an electrically neutral solution as well. The default formulation is workable; its main defect is that the counter-ions that should be in the diffuse layer are retained in the solution. The model results are adequate, provided solutions and surfaces are not separated or the exact concentrations of aqueous counter-ions are not critical to the investigation.
The
-diffuse_layer
and
-Donnan
identifiers activate models to balance the accumulation of surface charge with an explicit calculation of the diffuse-layer composition. When these identifiers are used, the composition of the diffuse layer is calculated and an additional printout of the elemental composition of the diffuse layer is produced. The
-diffuse_layer
identifier calculates the moles of each aqueous species in the diffuse layer according to the method of Borkovec and Westall (1983) and the assumption that the diffuse layer is a constant thickness (optionally input with
-diffuse_layer
, default is 10
-8
m). The variation of thickness of the diffuse layer with ionic strength is ignored. The net charge in the diffuse layer exactly balances the net surface charge. The
-diffuse_layer
calculation requires an integration that is slow and liable to failure under certain conditions. The
-Donnan
calculation is much faster and more robust, and gives results that are usually similar to the
-diffuse_layer
calculation.
In the
-Donnan
calculation, the concentrations in the diffuse layer are averaged and computed with:
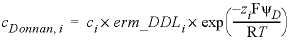 , (7)
, (7)
where cDonnan, i is the concentration of species i in the Donnan pore space (mol/L), ci is the concentration in the free (uncharged) solution, erm_DDLi is an enrichment factor (unitless) that can be defined in keyword SOLUTION_SPECIES, and zi is the charge number (unitless). The potential ψD is adjusted to let the charge of the Donnan volume counterbalance the surface charge:
 , (8)
, (8)
where σsurface is the surface charge (eq/L).
Conceptually, the results of using the explicit diffuse-layer calculations are correct--charge imbalances on the surface are balanced in the diffuse layer and the solution remains charge balanced. Great uncertainties exist in the true composition of the diffuse layer and the thickness of the diffuse layer. The ion complexation in the bulk solution is assumed to apply in the diffuse layer, which is unlikely because of changes in the dielectric constant of water near the charged surface. Identifier
-erm_ddl
in keyword SOLUTION_SPECIES can correct for such effects if needed, but it is a primitive and arbitrary correction. The explicit diffuse layer calculation is based on assumptions that allow the volume of water in the diffuse layer to remain small relative to the solution volume. It is possible, especially for solutions of low ionic strength, for the calculated concentration of an element to be negative in the integrated diffuse layer (calculated with identifier
-diffuse_layer
). In this case, the assumed thickness of the diffuse layer is too small (or perhaps the entire diffuse-layer approach is inappropriate) and the program stops with an error message. The identifier
-only_counter_ions
offers an option to let only the counter-ions increase in concentration in the diffuse layer, and to leave the co-ions at the same concentration in the diffuse layer as in the bulk solution. The counter-ions have a higher concentration in the diffuse layer than without this option, because co-ion exclusion is neglected. Alternatively, when using
-only_counter_ions and -Donnan
, the co-ion concentration is zero in the Donnan pore space. In this case, the counter-ions will have a smaller concentration in the Donnan layer with
-only_counter_ions true,
than with
-only_counter_ions
false.
A third alternative for modeling surface-complexation reactions, in addition to the default,
-diffuse_layer
, and
-cd_music
, is to ignore the surface potential entirely. The
-no_edl
identifier eliminates the potential term from mass-action expressions for surface species, eliminates any charge-balance equations for surfaces, and eliminates any charge-potential relationships. The charge on the surface is calculated and saved with the surface composition, and an equal and opposite charge is stored with the aqueous phase. All of the cautions about separation of charge, mentioned in the previous paragraphs, apply to the calculation using
-no_edl
. No explicit calculation of the diffuse-layer composition is available when using
-no_edl
.
For transport calculations, it is much faster in terms of CPU time to use either the default (no explicit diffuse layer calculation) or
-no_edl
. However,
-Donnan
and
-diffuse_layer
can be used to test the sensitivity of the results to diffuse-layer effects.
After a set of batch-reaction calculations has been simulated, it is possible to save the resulting surface composition with the SAVE keyword. If the new composition is not saved, the surface composition will remain the same as it was before the batch-reaction calculations. After it has been defined or saved, the surface composition may be used in subsequent simulations through the USE keyword. By using the RUN_CELLS data block, the results of the batch-reaction calculations, including the surface-assemblage composition, are automatically saved. In
ADVECTION
and TRANSPORT simulations, the surface assemblages in the column are automatically saved after each shift.
Example data block 2
Line 0d: SURFACE 1 Neutral surface composition
Line 1: Surf_wOH 0.3 660. 0.25
Line 1a: Surf_sOH 0.003
Line 2: Surfc_sOH Fe(OH)3(a) equilibrium_phase 0.001
Line 2b: Surfd_sOH Al(OH)3(a) kinetic_reactant 0.001
Explanation 2
Line 0d:
SURFACE
[
number
] [
description
]
Same as Example data block 1.
Line 1:
surface binding-site formula,
(
sites
or
site density
)
, specific_area_per_gram, grams,
[
Dw
coefficient
]
surface binding-site formula
--Formula for a surface that is charge balanced.
sites
--Total number of sites for this binding site, in moles; applies when
-sites_units
is
absolute
.
site density
--Site density for this binding site, in sites per square nanometer; applies when
-sites_units
is
density
.
specific_area_per_gram
--Specific area of surface, in m
2
/g (square meter per gram). Default is 600 m
2
/g.
grams
--Mass of solid for calculation of surface area, g (gram); surface area is
grams
times
specific_area_per_gram
. Default is 0 g.
Dw
coefficient
--Optional diffusion coefficient for the surface, m
2
/s; applies only when
-multi_D
is true in a TRANSPORT calculation. If coefficient > 0, the surface is transported as a colloid with advective, dispersive, and diffusive transport. Default is 0 m
2
/s, which means the surface is immobile.
Line 2:
surface binding-site formula, name,
[(
equilibrium_phase
or
kinetic_reactant
)]
, sites_per_mole, specific_area_per_mole
Same as Line 9 in Example data block 1.
Notes 2
The difference between Example data block 2 and Example data block 1 is that no initial surface-composition calculation is performed in Example data block 2. The composition of the surface assemblage must be given precisely (from chemical analysis) and charge-balanced to avoid spurious pH and redox reactions. Additional surfaces and binding sites can be defined by repeating Lines 2 and 9 from Example data block 1. All other identifiers listed for Example data block 1 also can be included.
Example problems
The keyword
SURFACE
is used in example problems 8, 14, 19, and 21.
| Next || Previous || Top |